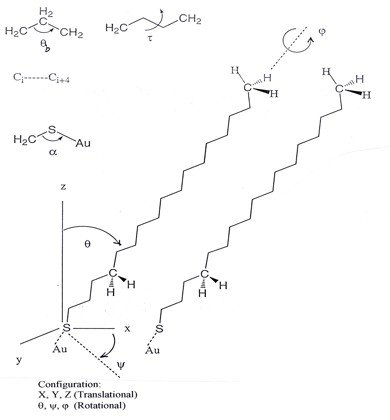
Molecular Model
In order to generate the stress-strain relationship and eventually calculate the values of the mechanical properties of a material, the total force, F, must be estimated beforehand. This is done through the computation of the total energy, E, of the system as F is defined as the gradient of E according to classical mechanics [1]. The potential energy, E, of the molecular system in question, namely, a SAM, depends upon the positions in space, denoted here as , of all atoms,
where n is the total number of atoms. The mathematical form (equation above) of the potential energy function consists of two terms, namely, bonded and non-bonded. The bonded terms describe contributions from atoms which are covalently bound, and are energy functions for bond stretching , angle bending , torsional angles , and out-of-plane bends , respectively. The non-bonded terms represent contributions to potential energy coming from interactions between atoms that are not covalently bound, and include van der Waals and electrostatic interactions. All energy terms depend upon the Cartesian/configurational and internal coordinates of the atoms.
- R. Henda (2004). Mechanical Properties of Self-assembled Organic Monolayers: Experimental Techniques and Modelling Approaches, Chapter 10, in Applied Scanning Probe Methods (NanoScience & Technol. series), Bhushan, B. (USA), Fuchs, H. (Germany), and Hosaka, S. (Japan) (Eds.), Springer-Verlag, New York/Heidelberg, pp. 303-326, ISBN 3-540-00527-7.