Etomica Modules
This page provides access to a suite of instructional modules based in molecular simulation. Their purpose is to foster molecular understanding of phenomena and processes commonly taught in standard science and engineering courses. They are interactive, graphically oriented, and quantitative (i.e., they provide real data for the models being simulated). Instructions, examples, and homework problems are provided as part of most modules; these resources may be accessed from the corresponding links in the Index of Molecular Simulation Modules, below.
The modules differ in the level of understanding needed to fully appreciate them, covering a range of topics across high-school, undergraduate, and graduate levels. However, they are all easy to run and control, and anyone with an interest in physics and chemistry should be able to learn from them.
This is a project of CACHE. Funding for this development has been provided by the National Science Foundation, Grants DUE-9752243 and DUE-0618521.
Download Instructions
The modules are distributed together as a single self-contained application that allows selecting and launching individual modules. Note that this distribution does not require Java to be installed on your system!
You can download the latest build from GitHub here and run the application from the zip archive. On MacOS and Windows your system may need to be configured to allow running unsigned applications.
Key Bindings
For most of the modules, some of the keys on your keyboard can be used to manipulate the configuration display.
First, make sure the focus is on the configuration display (select the Configuration tab and click somewhere in the display).
Then, keypresses will perform the actions as follows:
- The 1 key causes display of the first shell of periodic images (2 does the 2nd as well, etc.); 0 returns to just the original central image. Also, in 3D, the i key will toggle between two displays of image shells.
- The b key ("boundary") affects the presentation of the boundary boxes when multiple image shells are displayed. In 3D, it cycles through several options when pressed multiple times; in 2D there are only two such options, but it takes several presses to cycle through them.
- The o key will toggle drawing of "overlap images", i.e., parts of images that fall in a neighboring box. This applies only if periodic images are displayed. The effect is rather subtle (2D only).
- +/- keys (without shift key) zooms in/out (3D only).
- The x,y,z keys will rotate about respective axes (3D only).
- The h key ("home") will present the view in the standard orientation and size.
- The p key toggles the perspective on/off (3D only).
- Arrow keys will move the image left/right/up/down (3D only).
Remember to have the focus on the configuration when applying these!
Additionally, the mouse can be used to manipulate 3D images:
- Click on the image and drag to rotate.
- Hold the Shift key and click while moving the mouse vertically changes the zoom level.
Index of Molecular Simulation Modules
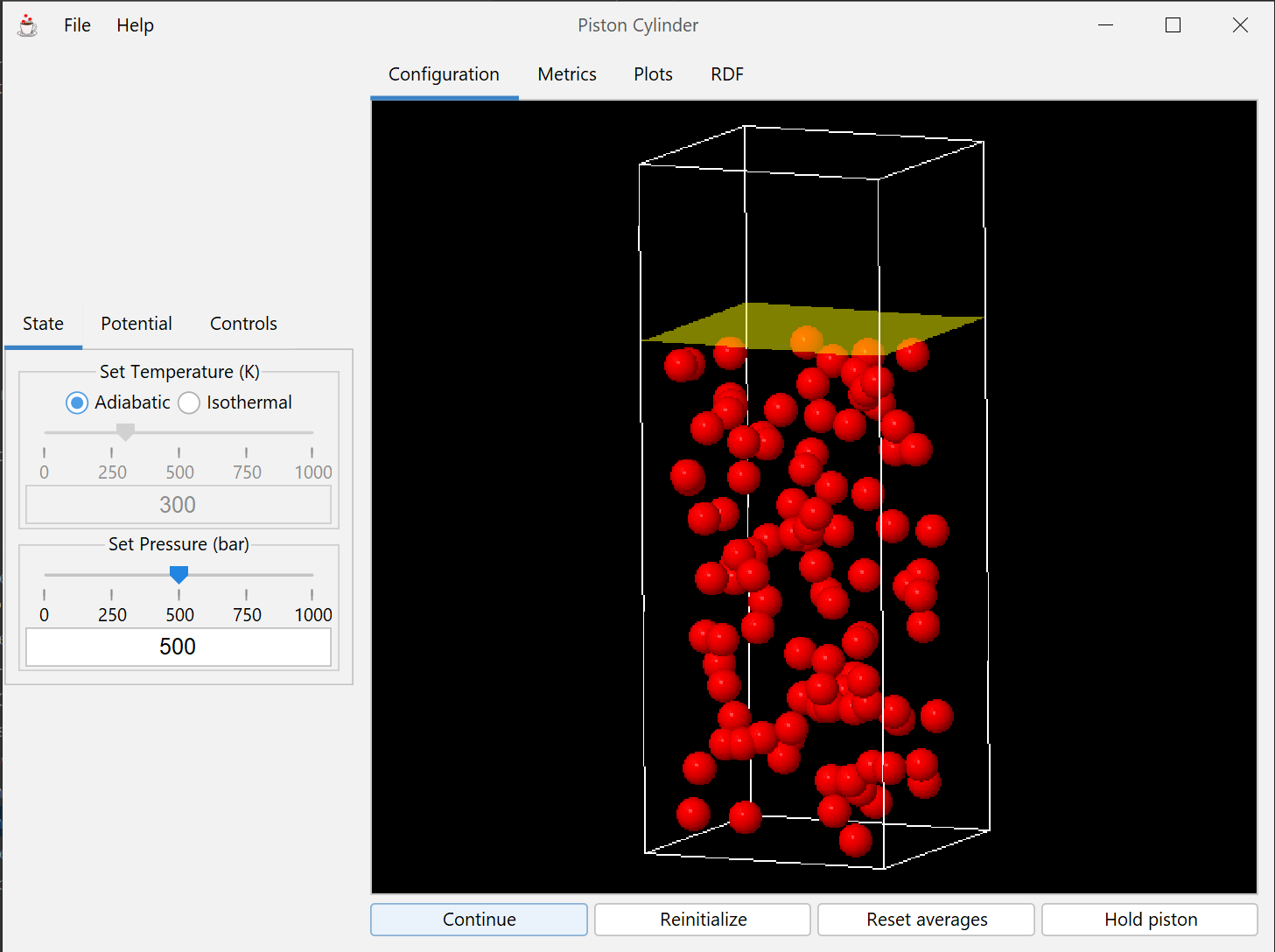
The piston-cylinder apparatus is a standard tool used to illustrate thermodynamic concepts involving heat, work, and internal energy. This module simulates a collection of atoms in a chamber with a movable wall under external pressure.
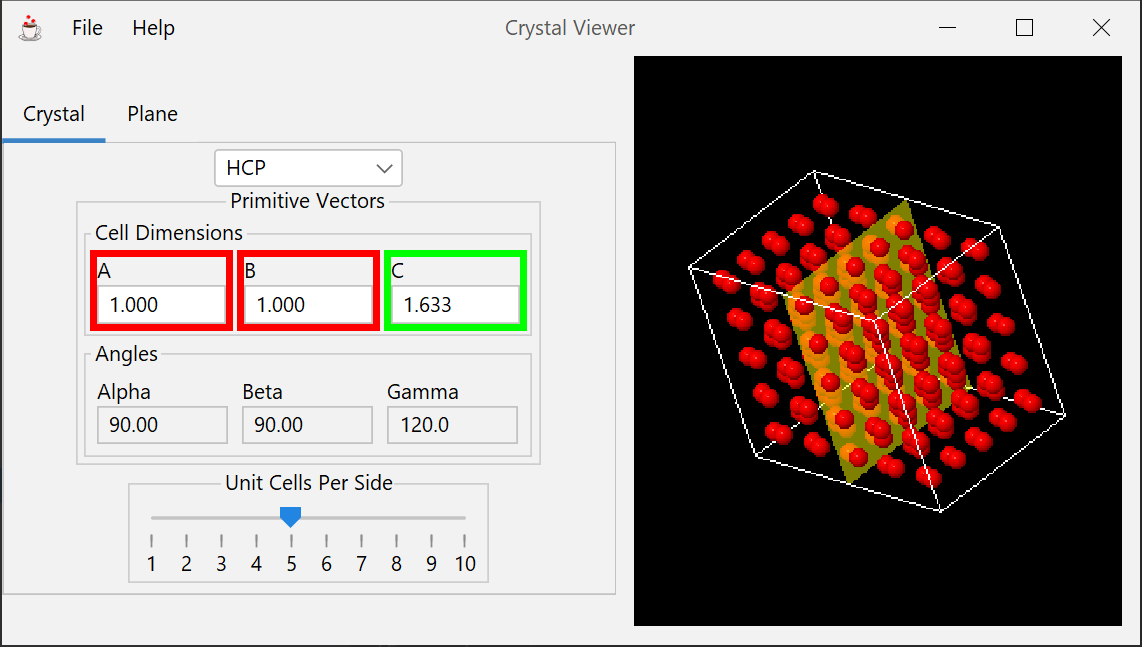
Permits viewing of static 3-dimensional lattices and the planes they define. Various elementary lattices can be selected, and any plane or surface can be viewed by specifying it via its Miller indices. Image can be rotated to permit viewing from any angle.
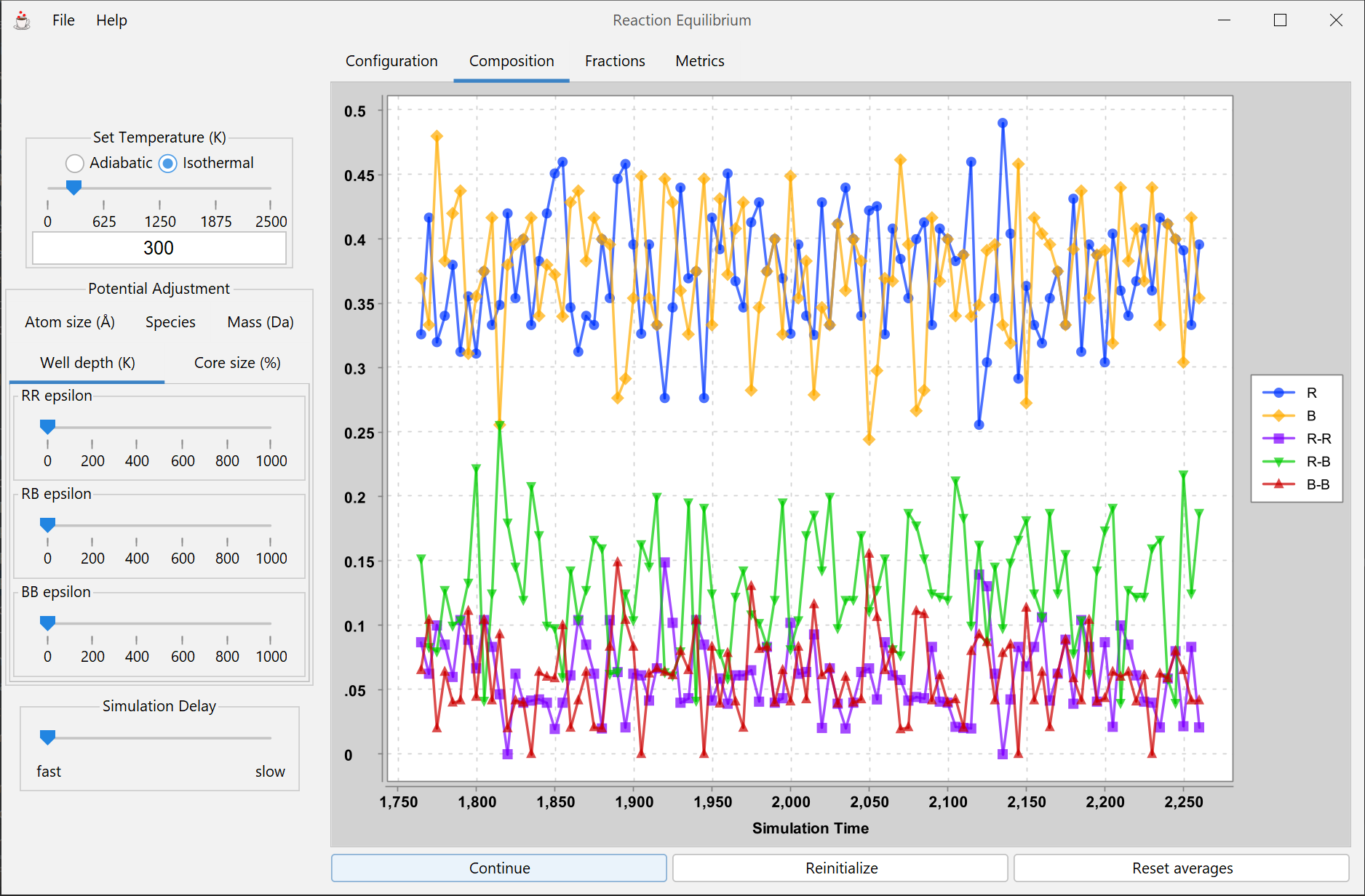
Simple reaction equlibrium involving two atomic species and the three dimeric molecules they can form. Atoms move about via 2-D molecular dynamics, and can "react" to form dimers. Dynamic equilibria is demonstrated through constant recombining of atoms, and equilibria can be quantified and analyzed with thermodynamic reaction equilibria models.
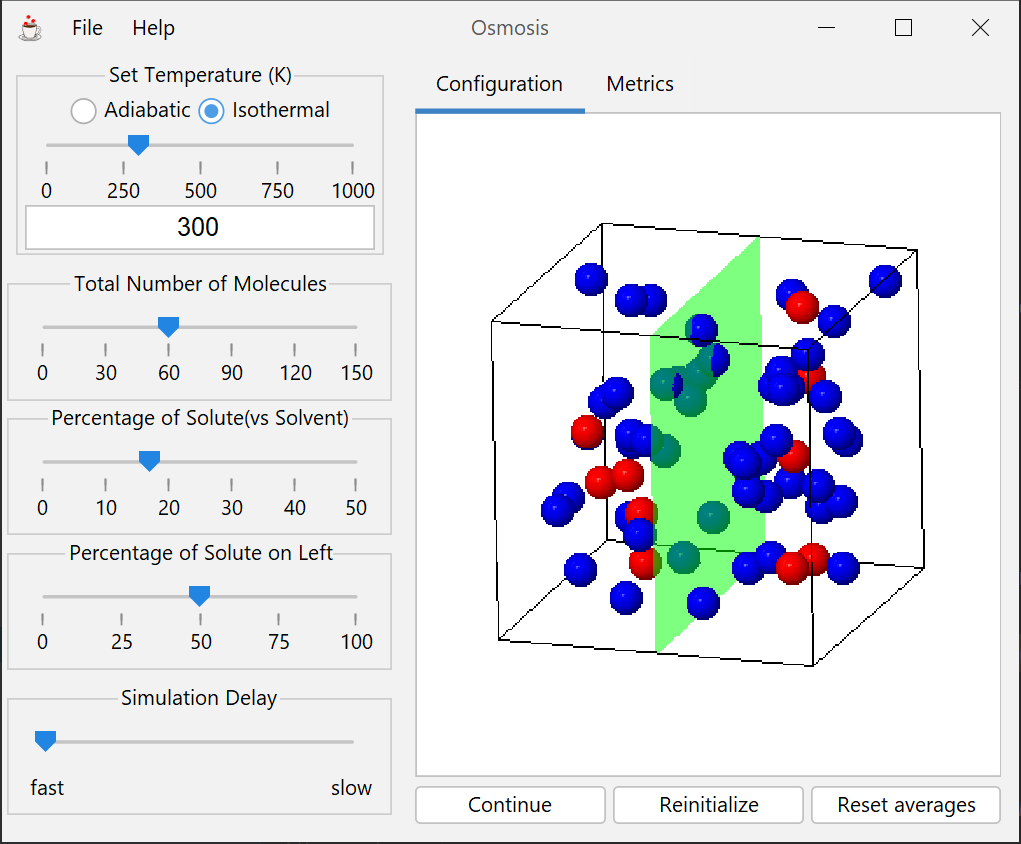
Simulation of a system of two species and a semipermeable membrane. Allows measurement of the osmotic pressure (the difference in pressure between the phases on each side of the membrane) as a function of density and mixture mole fraction.
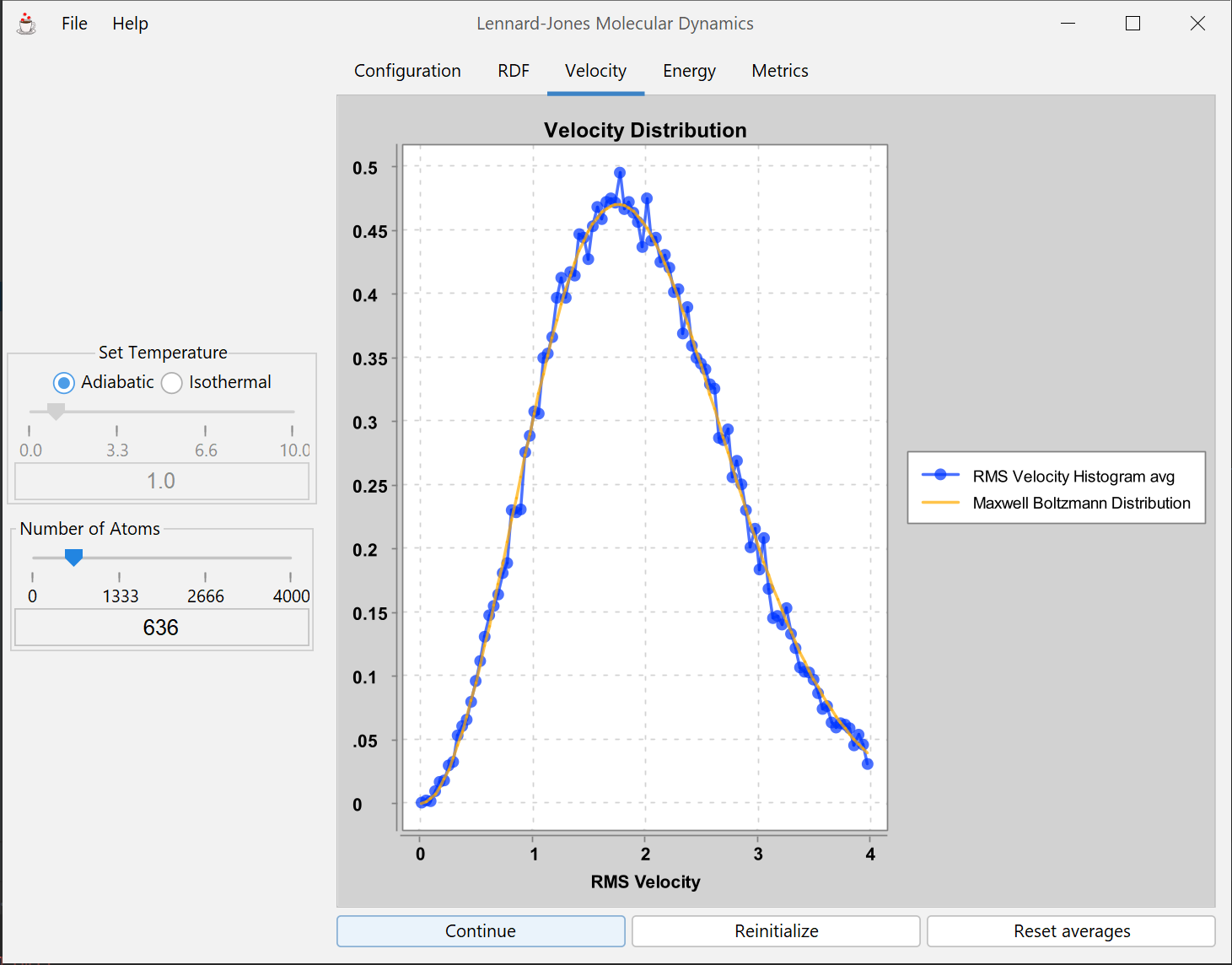
Molecular dynamics of a 2-dimensional mono-atomic Lennard-Jones system. The Lennard-Jones model is a simple but widely-used approximation for the way atoms interact. Elementary molecular features of this model's dynamical and structural behavior are calculated in this simulation. More appropriate for a graduate-level student.
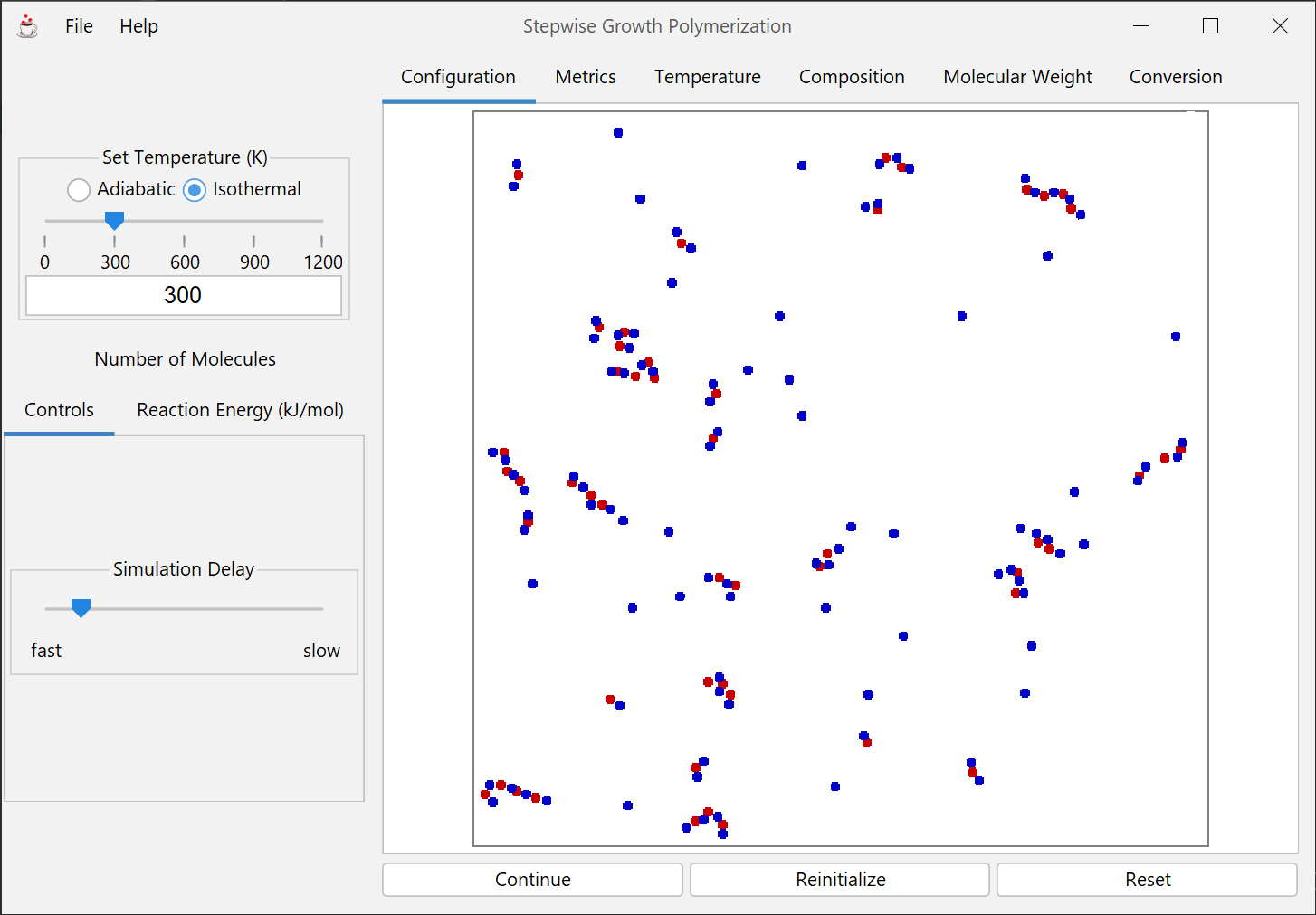
Model of two fundamental polymerization reaction types: stepwise growth and chain addition. The difference between the reaction mechanisms can be visualized, the resulting products of each can be observed, and calculations of molecular weights, molecular weight distributions, and kinetics can be conducted.
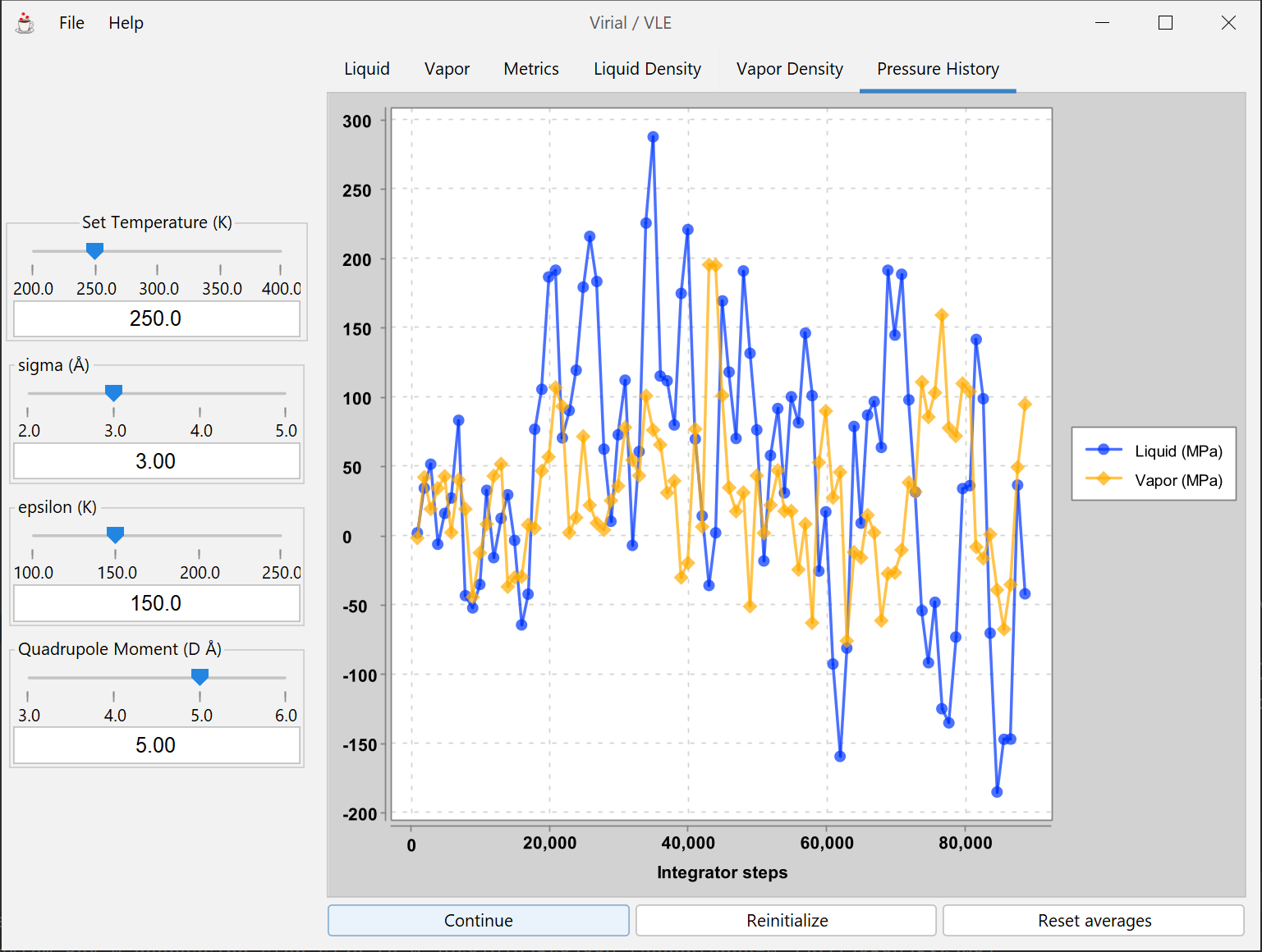
A molecular model is fit to experimental data for the 2nd virial coefficient, and the vapor-liquid equilibrium (VLE) behavior of the fitted model is examined by Gibbs-ensemble molecular simulation. Resulting data can be compared to experimental VLE data for the system used to fit the model.
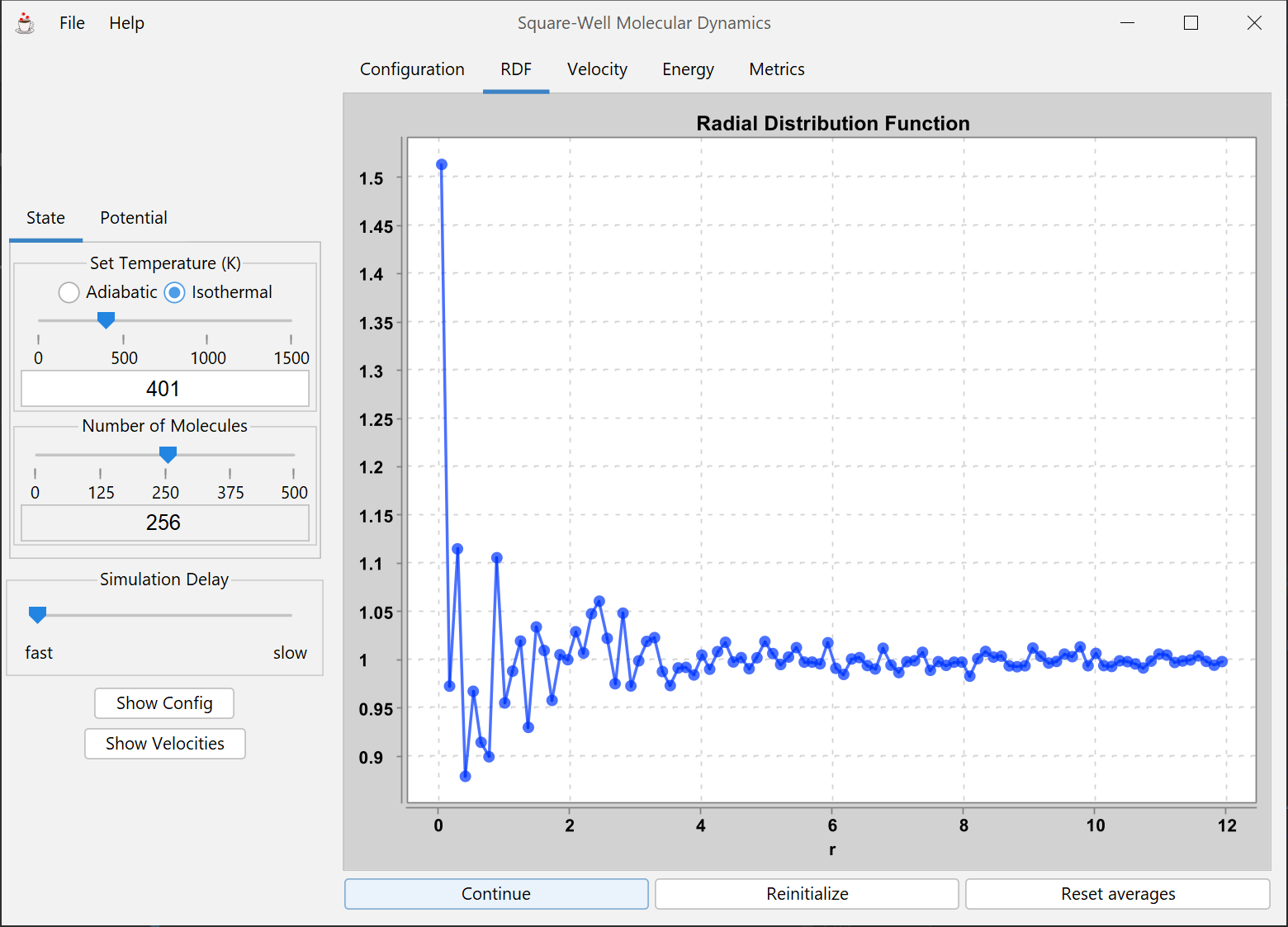
An integrated tutorial on pressure, temperature, density, and model equations through the piston/cylinder module combined with 2D and 3D simulations using periodic boundary conditions. The simulations are based on the square well potential, which characterizes the fundamental repulsions and attractions in a discontinuous but simple manner.
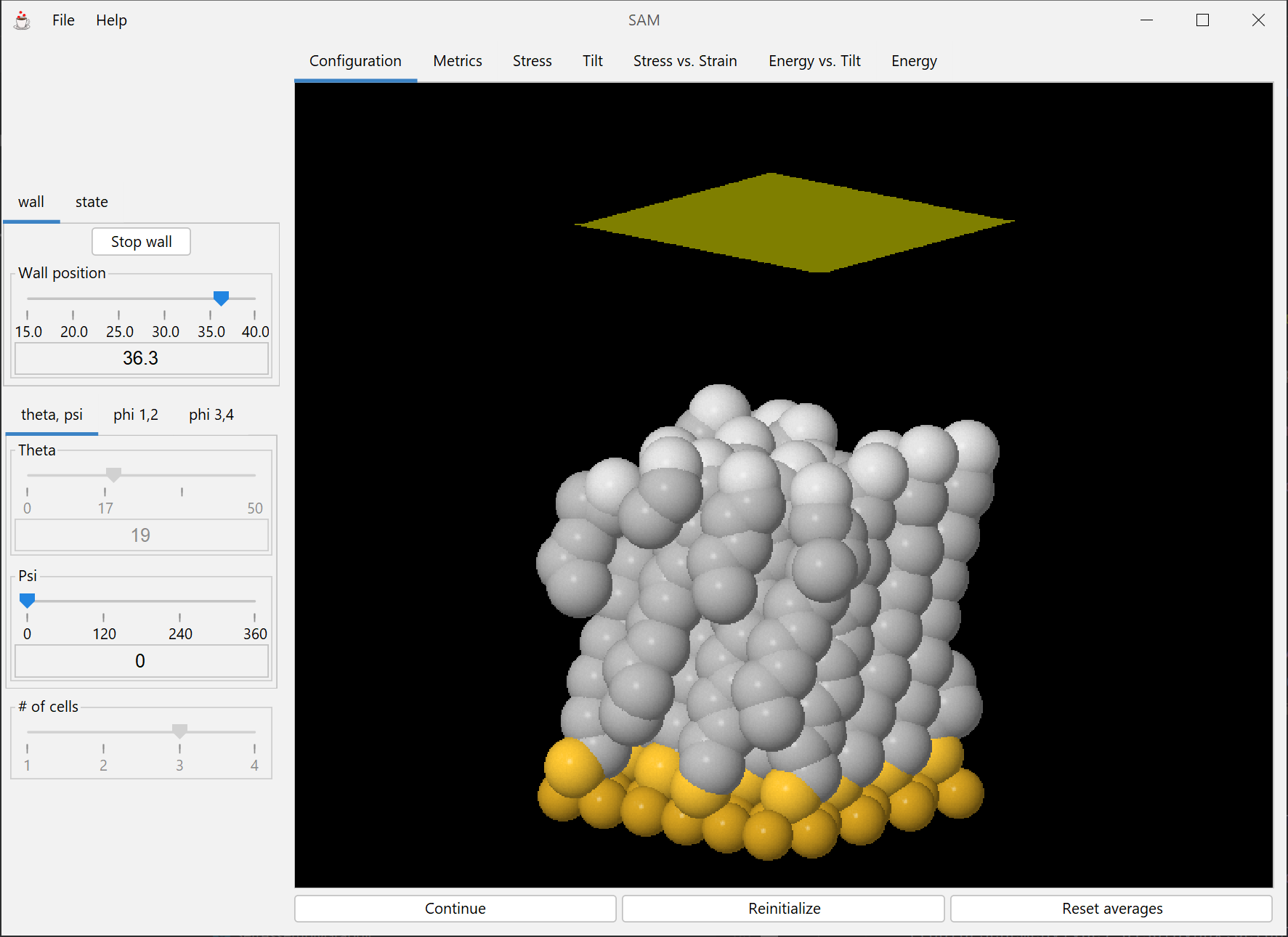
Demonstration of mechanical properties of tethered monolayers. The module can be used as a supplement in materials science and engineering courses to illustrate the molecular basis of stress and strain.
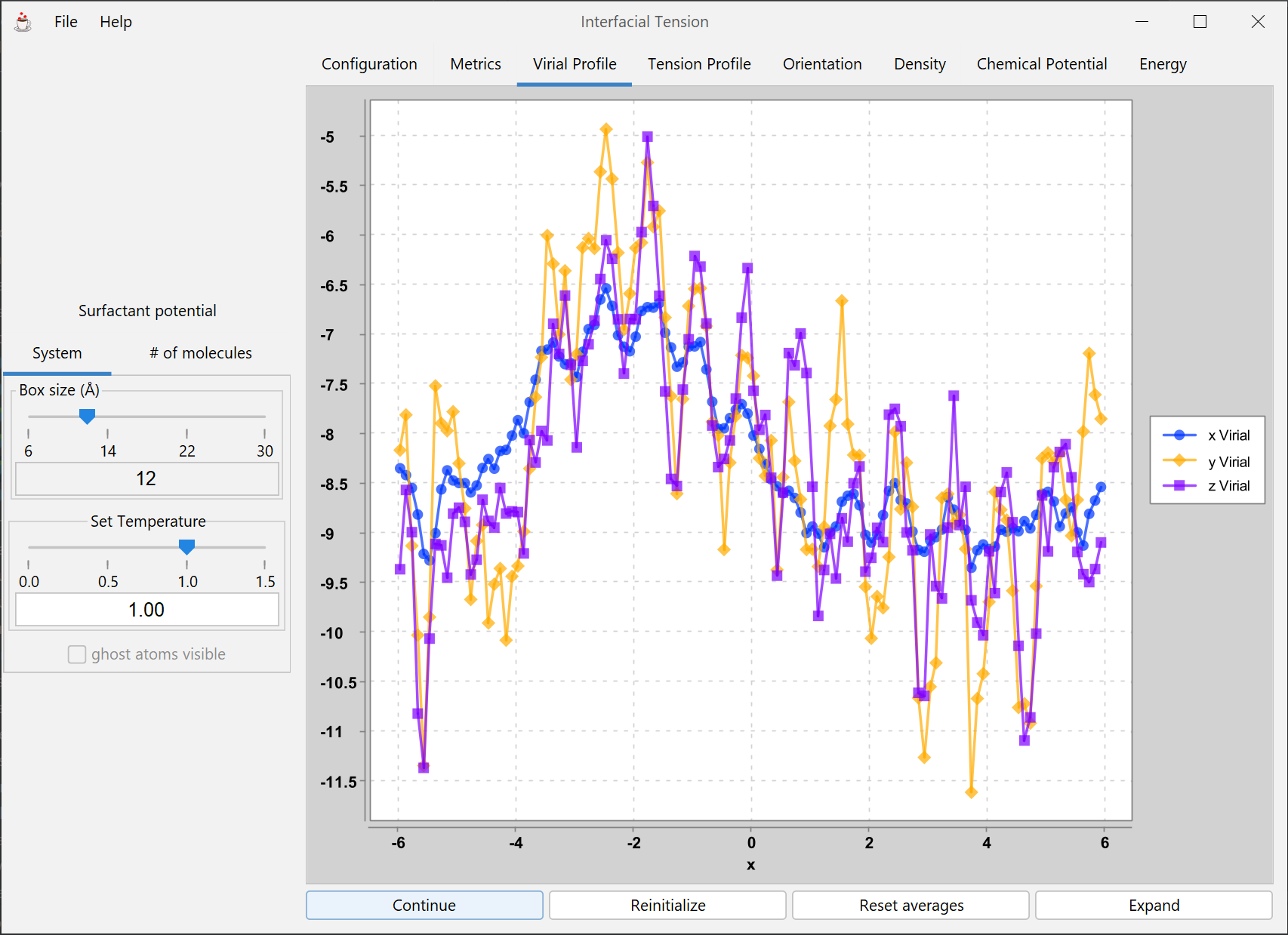
Model of an explicit vapor-liquid interface, permitting computation of the surface tension and other thermodynamic and structural properties at various conditions.
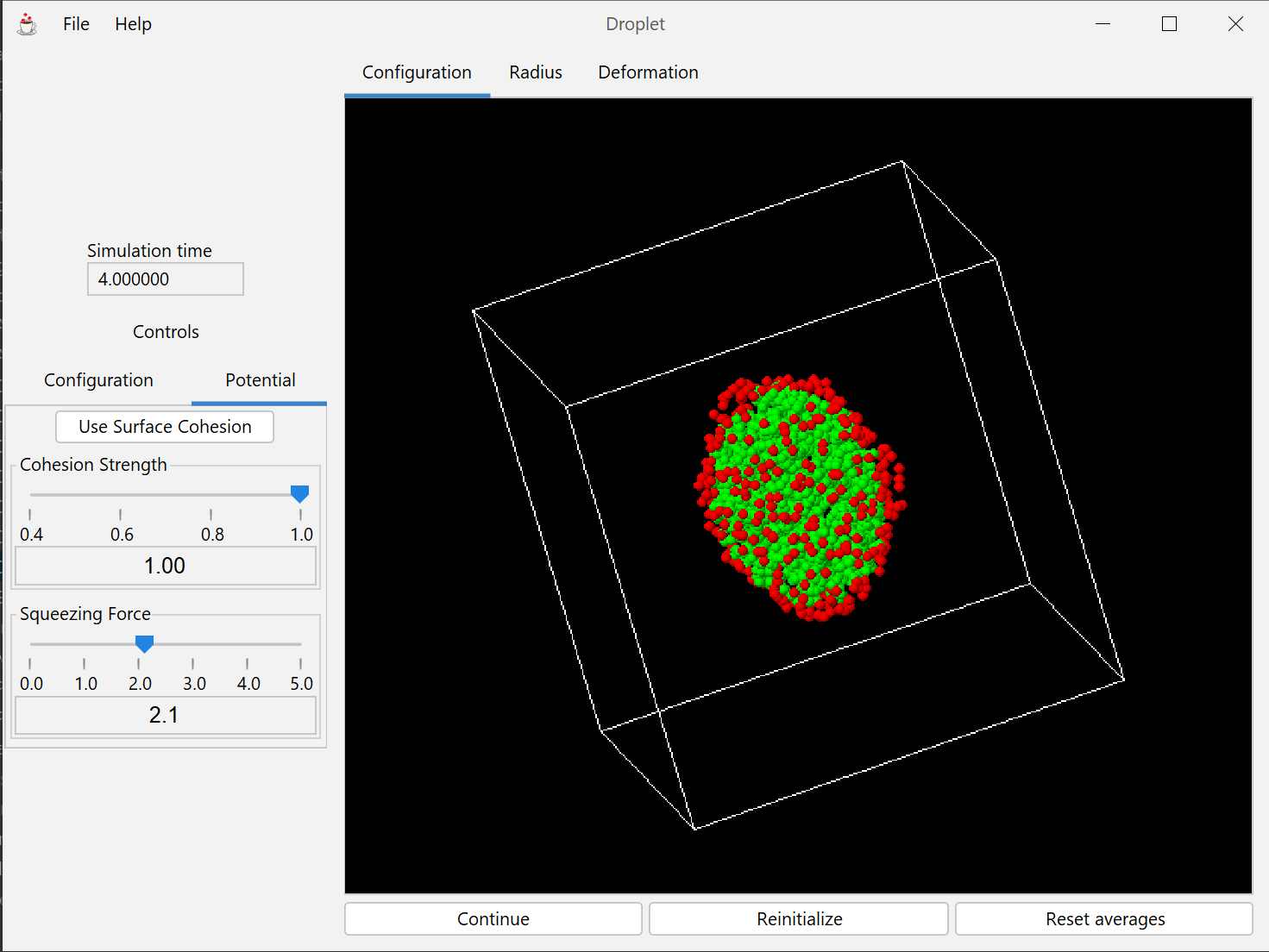
This module discusses curvature of the surface of a liquid drop, and also liquid-liquid interfacial tension.
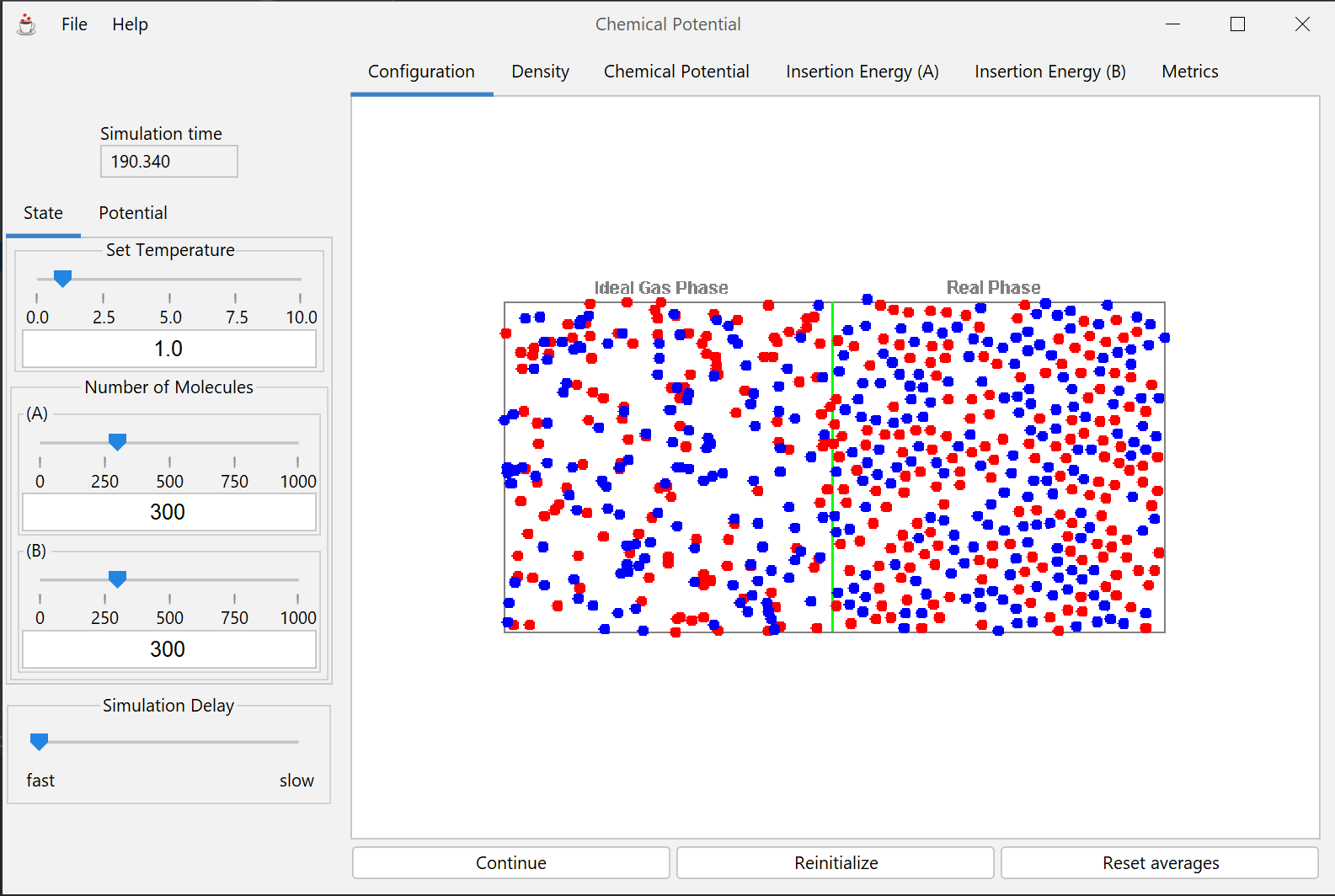
This module presents an interactive molecular simulation that shows how diffusion is related to the chemical potential, and how the chemical potential depends on factors such as concentration, pressure, temperature and molecular properties.
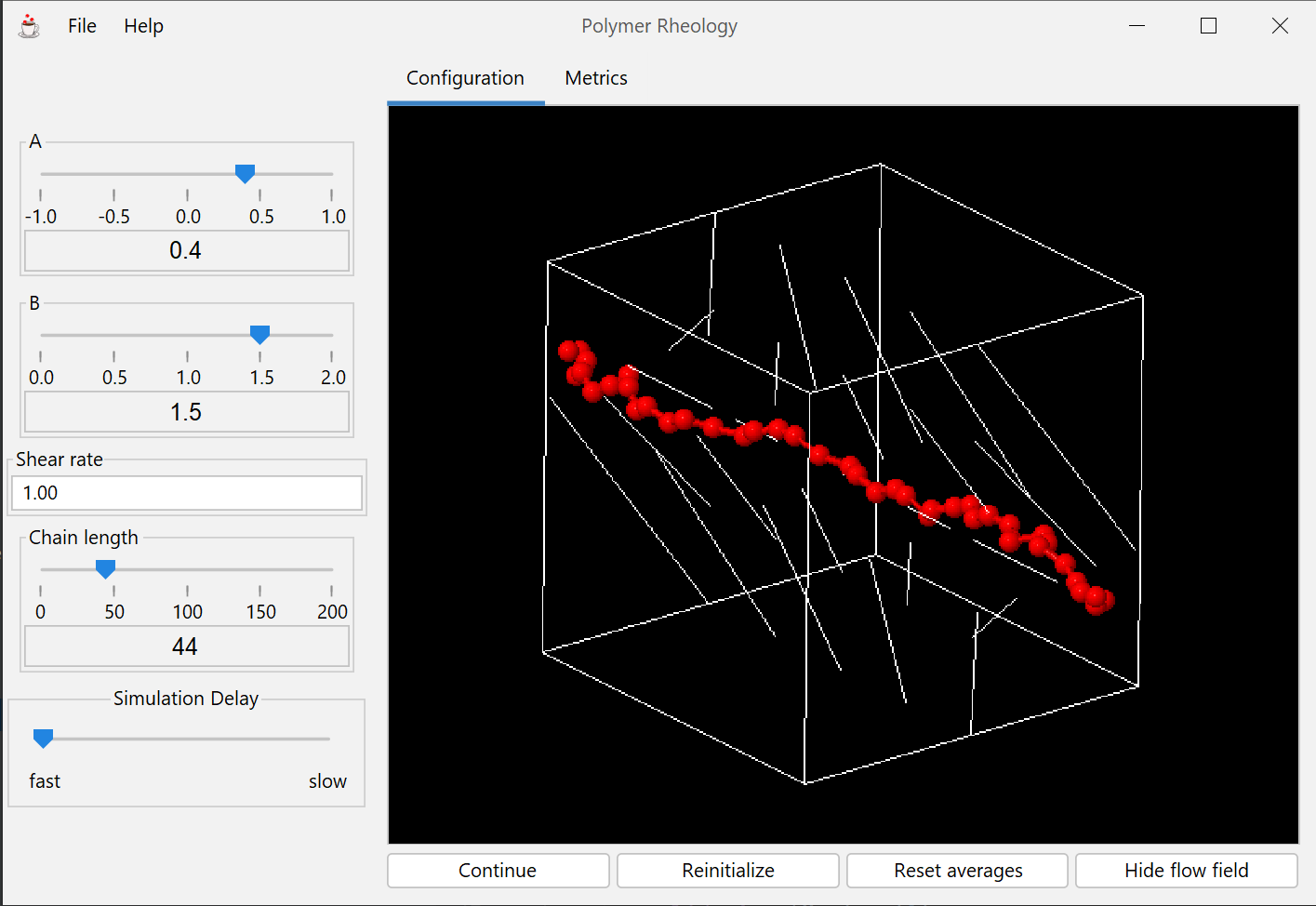
This module presents a Brownian dynamics simulation of a polymer in which the user may interact by adjusting important model parameters and flow conditions.
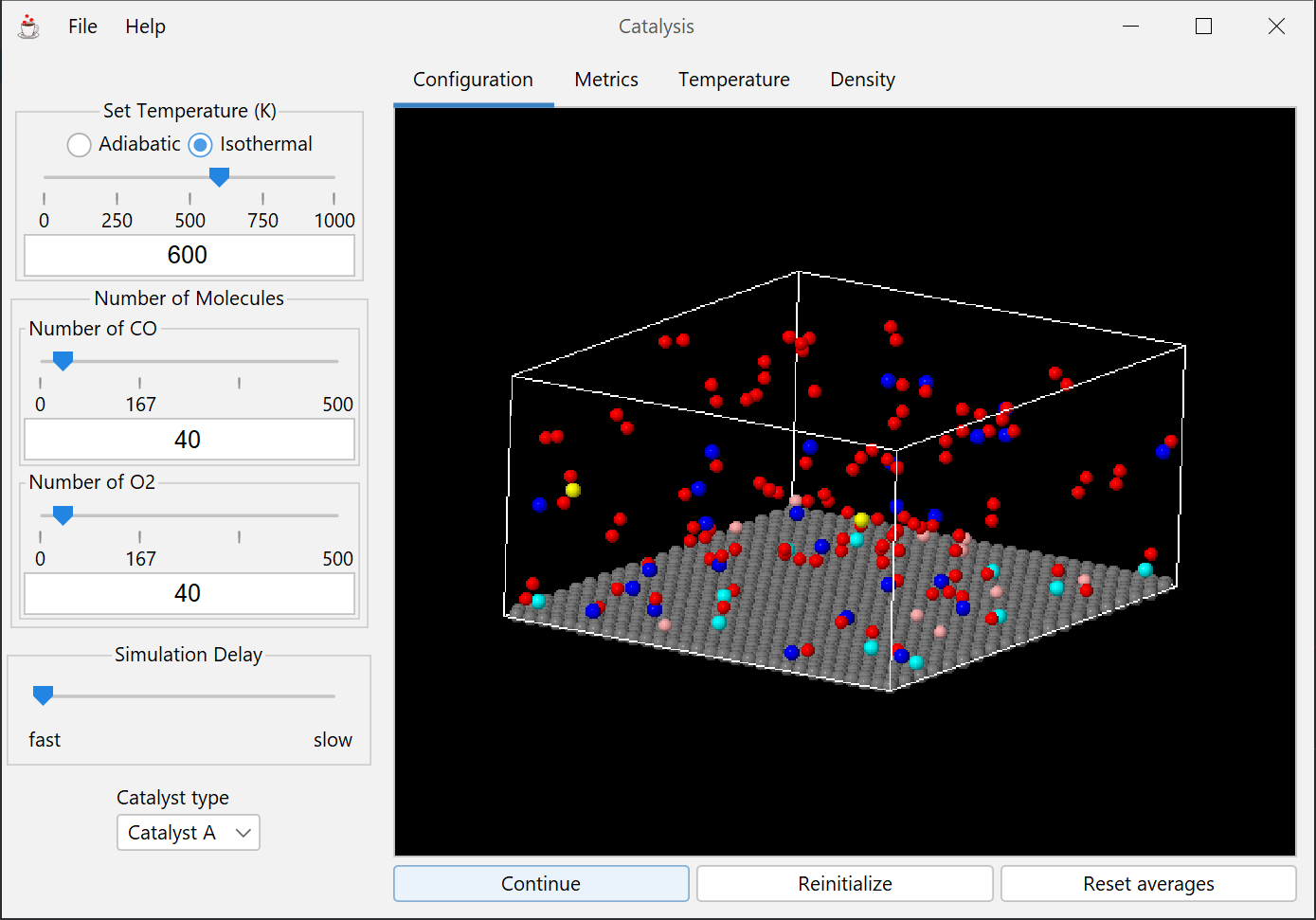
Demonstrates the dynamical molecular-level interactions and events relevant to heterogeneous catalysis, and provides a framework for applying catalytic reaction engineering principles.
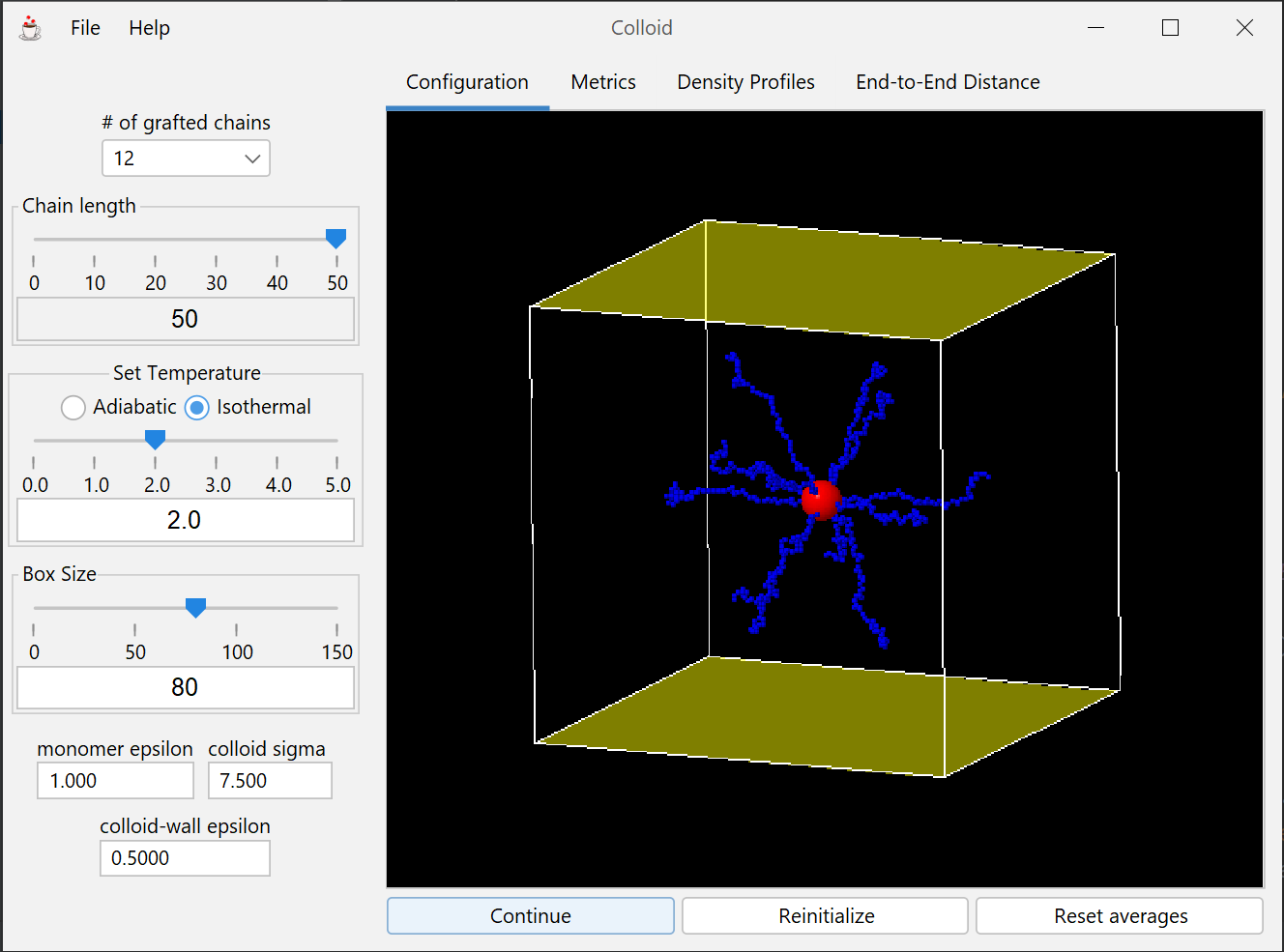
This module shows that colloid brush-surface interactions are dictated by a balance between entropic and enthalpic phenomena due to the grafted polymer chains.
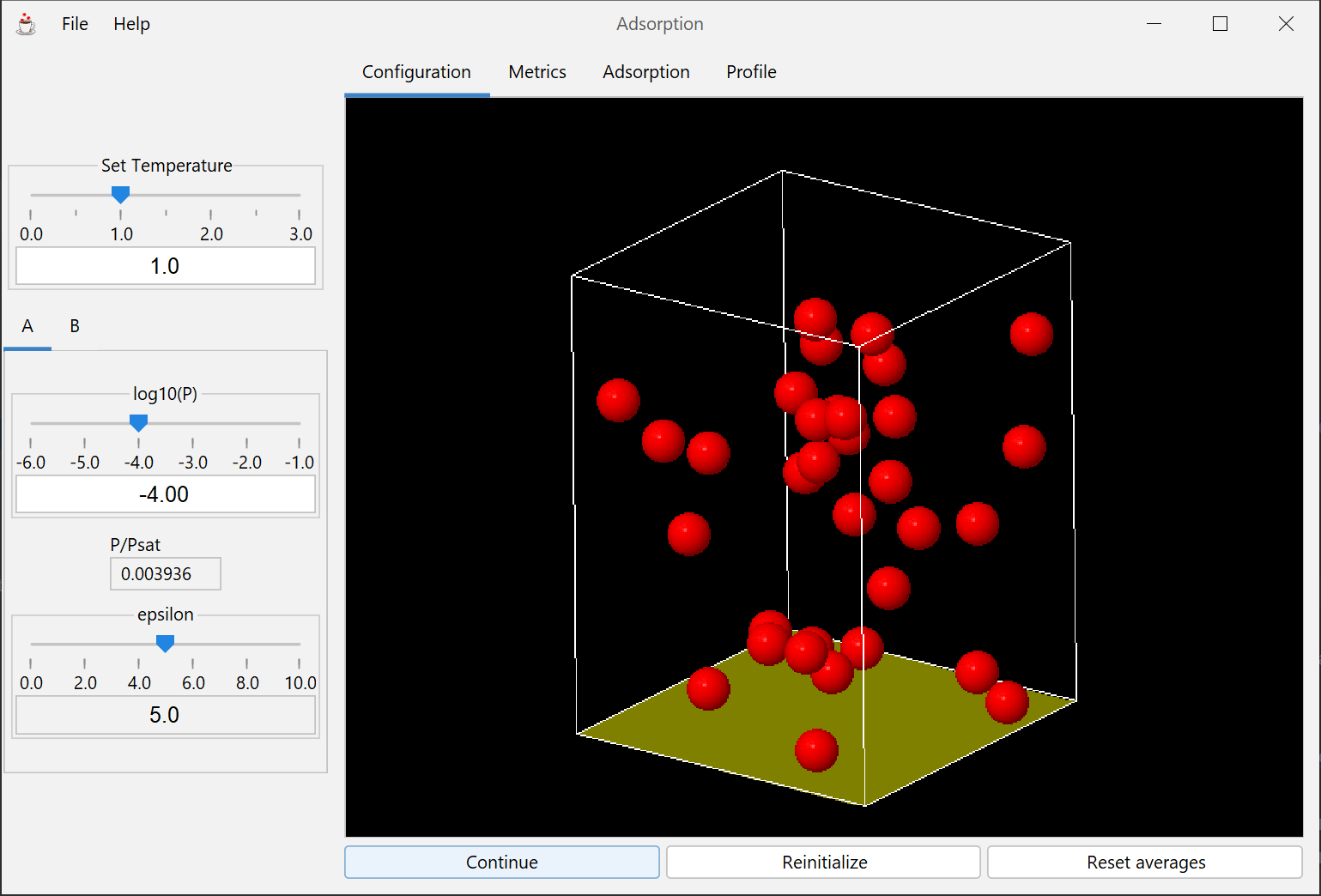
This module explores the thermodynamics of adsorption through molecular simulation. The principal focus is on physisorption and its applications, though many of the same models and concepts are also applicable to chemisorption.
Dual Control Volume Grand-Canonical Molecular Dynamics (DCVGCMD) simulation, in which a long simulation volume is subject to grand-canonical Monte Carlo at opposite ends, with molecular dynamics in between. The MC simulations establish a chemical-potential gradient, and the resulting diffusion process can be used to measure the diffusion coefficient.
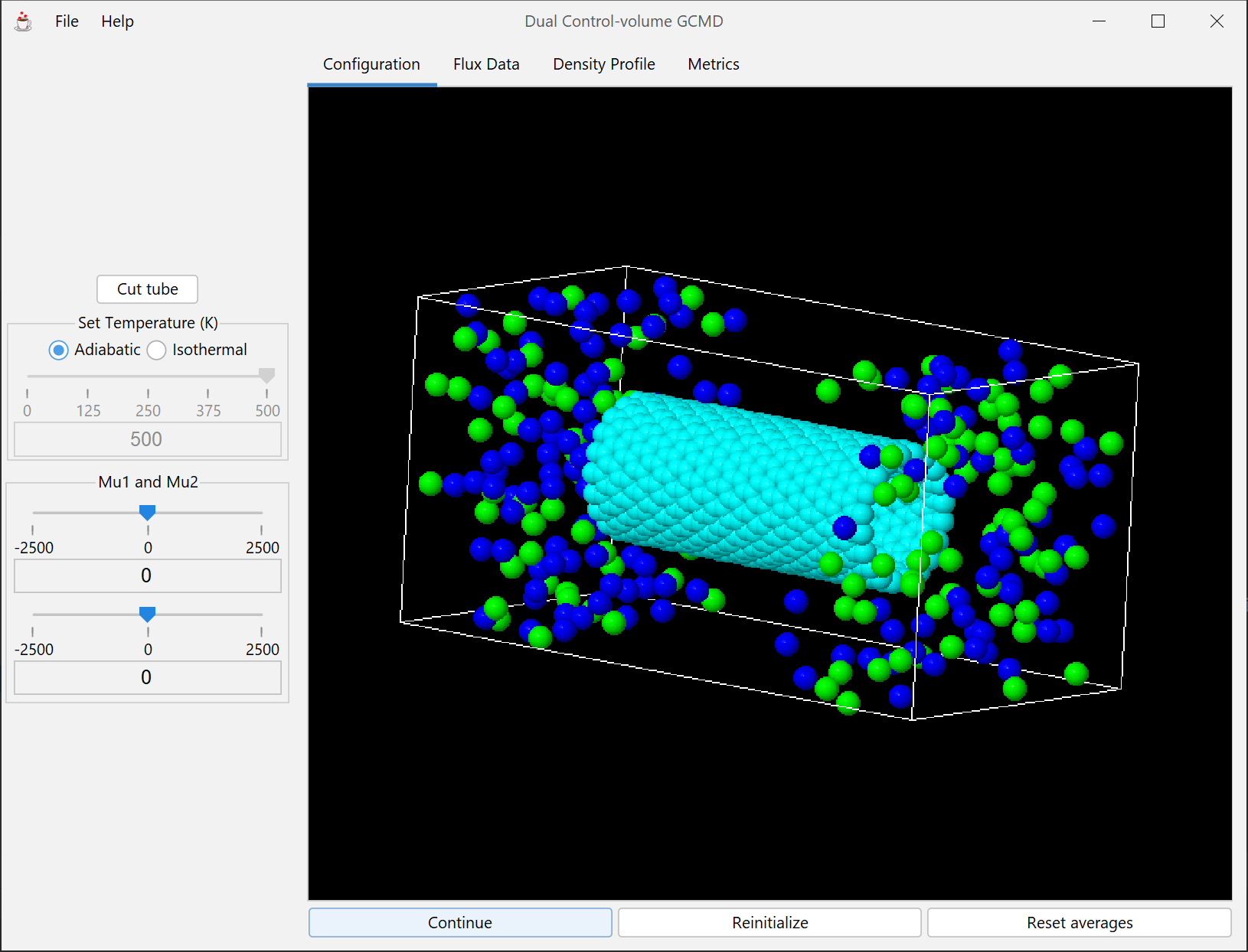
An extension of the previous DCVGCMD module. The atoms must diffuse through a tube composed of hexagonally arranged atoms.
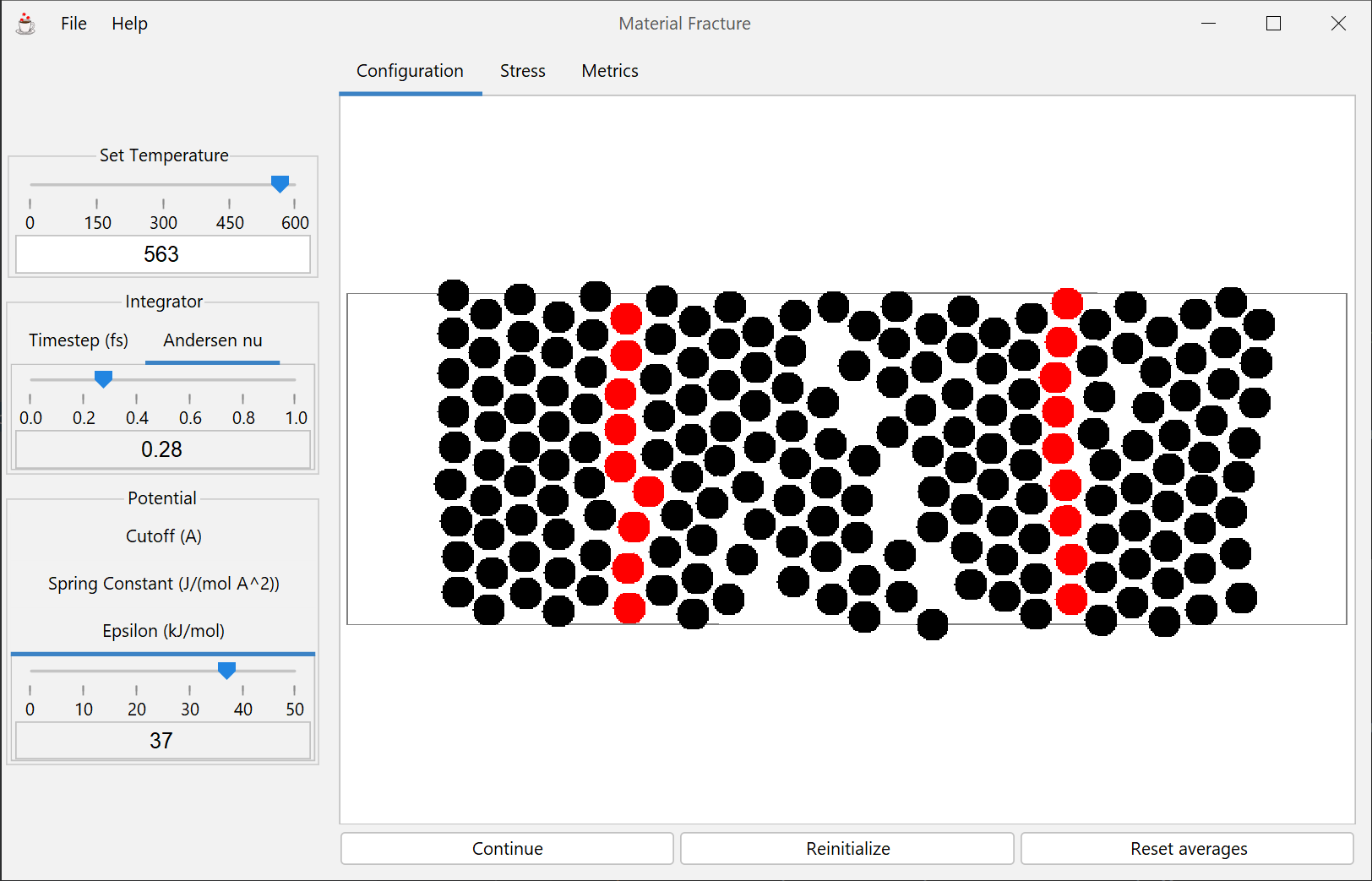
2-D simulation of a monatomic species in the solid phase. Stress is applied and the resulting strain may be observed to and beyond the point of fracture. Vacancy defects may be introduced to examine their effect on the material's strength.
Demonstrates the independent collective motions that can be used to describe the dynamics of a system of one-dimensional coupled harmonic oscillators.